
scRNA-seq 的原始數據格式和目前大多數 scRNA-seq 分析過程都基于 FASTQ 文件(或壓縮格式 fq.gz)。Illumina 平臺測序數據默認生成 BCL 格式文件,可以通過 CellRanger mkfastq 進行轉換。scRNAseq 的分析流程包括數據預處理、處理和擴展下游分析(圖 4),其中數據預處理包括質控、read 比對和表達量化;數據處理包括標準化、批次效應校正、歸一化、特征選擇(HVG 選擇)、降維與聚類、細胞分型注釋、差異表達分析(DEGs)、可視化;擴展下游分析包括擬時序、細胞間相互作用(CCI)、通路富集分析、基因調控網絡(GRN)等下游分析。整體來看,scRNAseq 分析方法層出不窮,沒有絕對完美適用于所有場景的方法,分析工具重要的是獲取生物學信息,難點在于選擇最合適的方法。本文中,我們將提出總結常見的單細胞轉錄組分析方法,并對其優缺點和適用范圍提出建議。
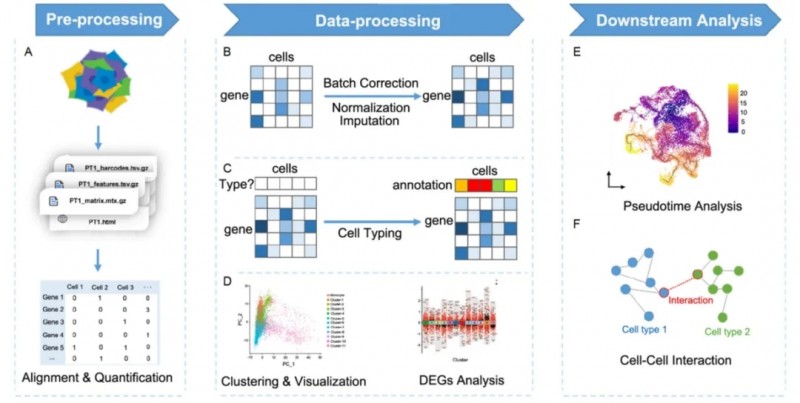
圖 4:單細胞分析概覽。A. 在預處理階段,基于測序數據,細胞 - 基因矩陣讀數通過單細胞讀數校正和定量產生;B. 分析使用的高質量細胞矩陣通過原始的基因表達矩陣獲得,通過去批次效應矯正批次,通過標準化降低生物學差異,補充未檢測到的基因;C. 依照或不依照先前的參考信息對細胞類型進行注釋;D. 轉錄組特征相似的細胞被歸為一類,稱為“細胞簇(cluster)”,細胞的可視化通過降維方法實現,差異基因分析對組間差異進行檢驗;E. 擬時序分析重建細胞轉錄水平變化的動力學過程;F. 細胞間轉錄組調控關系可以通過胞間互作分析進行推斷。
數據預處理
將原始測序數據通過濾除低質量 reads 和環境干擾與參考基因組進行比對和量化。從而得到每個細胞的特征計數矩陣和記錄其他信息的輔助文件,用于下游的數據分析(圖 4 A)。
1、質控
由于測序儀器問題、人為操作、細胞自發情況,或存在空液滴、雙細胞、死細胞等,不可避免地會產生低質量的測序數據(Chen 等,2019a ; Hao 等,2021b)。空液滴通常出現在液滴捕獲細胞外背景轉錄本而不是細胞時(Ilicic 等,2016 ; Kolodziejczyk 等,2015)。一種高度主觀的方法是根據曲線的膝點確定一個 UMI 閾值,并過濾掉 UMI 計數低的細胞。隨后使用 DropEst (Petukhov 等,2018)、EmptyDrops (Lun 等,2019) 和 DIEM (Alvarez 等,2020) 增強過濾效果。DropletQC (Muskovic and Powell, 2021) 量化未剪接前 mRNA 含量的核分數得分。MT 基因閾值雖然是衡量死細胞的標準,但它的選擇需要綜合考慮細胞生理因素 (Subramanian 等,2022)。近年來,基于深度學習的方法也應運而生,例如基于神經網絡的 EmptyNN (Yan 等,2021) 和基于深度生成模型的 CellBender (Fleming 等,2019),能夠有效識別空液滴中的背景轉錄本。
雙細胞是指兩個細胞包含在一個液滴中的情況,根據轉錄分布可分為同源雙峰和異源雙峰,均服從泊松統計量(Bloom, 2018)。絕大多數方法基于基因表達計算,利用先驗知識或深度學習獲取單峰與雙峰細胞的差異,然后訓練分類器進行篩選,例如基于最近鄰的 DoubletFinder (McGinnis 等,2019a)、Scrublet (Wolock 等,2019);基于反卷積的 DoubletDecon (DePasquale 等,2019)、基于變分自編碼器的 Solo (Bernstein 等,2020) 和基于集成算法的 Chord (Xiong 等,2021a)。此外,Scds 是另一種篩選方法,它依賴于基于共表達的雙聯體打分和基于二分類的雙聯體打分策略,實現 scRNA-seq 表達數據的雙聯體分離 (Bais and Kostka,2020)。一些方法使用其他特征,例如 demuxlet 它使用自然遺傳變異信息指導實驗并通過計算進行過濾 (Kang 等,2018)。
合理的質控需要綜合考慮技術性和生物性因素,這也是當前研究的主要方向。最近一種由生物數據驅動的自學習無監督質控方法 ddqc 被提出來,用于確定各種 GC 指標的具體閾值 (Macnair and Robinson,2023)。
2、reads 比對和定量
質控后剩余的高質量細胞需要將這些短 reads 映射到特定的參考基因組上進行比對,以此對基因表達水平進行定量。RNA 比對通常分為兩步:比對 reads 以建立索引和映射 RNA 剪接序列,前一步與 DNA reads 比對共用,解決錯配問題并設置索引參考;后一步是 RNA reads 比對所特有的,提供連通性信息。
早期二代測序結果是幾十對長度的堿基 reads。Seed-to-extend (Buhler,2001)(包括 MAQ (Li 等,2008a)、SOAP (Li 等,2008b)、CloudBurst (Schatz,2009)、ZOOM (Lin 等,2008))、BurrowsWheeler 變換方法 (Burrows and Wheeler,1994)(包括 SOAP2 (Li 等,2009)、Bowtie (Langmead 等,2009)、BWA (Li and Durbin,2009))、Needleman-Wunsch 方法(包括 Novocraft (Hercus,2009))和 suffix-tree 算法方法(包括 MUMmer 2 (Delcher 等,2002))都是百萬級短鏈 DNA 測序 reads 比對的有效工具。Bowtie 采用了一種依賴于 Burrows-Wheeler Transforming 的 FM-index 方法,如果 reads 有多個準確匹配則結果只報告一個,與 MAQ(Ferragina and Manzini,2001)相比,大大優化了運行內存和比對速度。BWA 是另一種基于 BWT 的比對方法,使用新的 SAM(Sequence Alignment/Map)格式輸出比對結果。基于 MAQ 和 Bowtie 兩種短鏈 DNA 比對算法,Cole Trapnell 于 2009 年提出了第一個針對 NGS 數據的 RNA-seq 比對方法 TopHat,它使用 2 -bit-per-base 編碼實現 reads 與哺乳動物基因組中剪接位點的有效比對,而無需事先知道剪接位點的具體信息(Trapnell 等,2009)。
上述方法在堿基對長度超過 50 bp 時比對精度急劇下降(Gupta 等,2018 ; Lebrigand 等,2020)。NGS 單細胞測序分析主要采用兩類方法:基于 Bowtie2 的方法和基于 seed 策略的方法(Langmead and Salzberg, 2012)。Bowtie2 是 Bowtie 的升級版,保留了 FM-index 依賴的 BWT 算法核心,允許有間隙比對,并使用單指令多數據(SIMD)擴展到長測序比對,同時提高運行速度。Daehwan Kim 在 Bowtie2 基礎上,先后提出了 TopHat2(Kim 等,2013)和 HISAT(Kim 等,2015)。種子策略主要有 STAR(Dobin 等,2013)和 Subread(Liao 等,2013)。STAR 基于最大可映射前綴(MMP)的思想,采用順序檢索的策略,將與參考匹配的最長部分 reads 設為種子 1,其余 read 繼續匹配,依次從種子 2 調用至種子 n。值得注意的是,Rsubread 完全基于 R 語言平臺實現了第一次 read 比對和基因量化的過程(Liao 等,2019)。
基因表達量化又可分為偽比對量化和基于 read 比對的量化。偽比對是指不采用上述嚴格的兩步法將所有 reads 比對到參考基因組上,包括選定的 k-mers 比對方法(Sailfish(Patro 等,2014)、Kallisto(Bray 等,2016)、Salmon(Patro 等,2017)、RapMap(Srivastava 等,2016)和 Barcode-UMI-Set (BUS) 比對方法 BUStools(Melsted 等,2019)。Kallisto-BUStools 是最新的工作流程,它使用 BUS 文件格式進行初始數據預處理,與 BUStools 一樣,偽比對結果和量化計數都保存在 BUS 文件中(Melsted 等,2021)。另一方面,基于 reads 比對的方法依賴于 RNA reads 比對方法的結果來量化基因。CellRanger 是 10x Genomic 公司指定替代 Longranger 的官方開源數據預處理軟件(Zheng 等,2017)。STARsolo 是替代 Cellranger 的 mapping/quantification 功能的工具,可實現多平臺測序數據的分析和基因表達之外的轉錄組特征的量化(Kaminow 等,2021)。其他基于 reads 比對的基因表達定量方法如 UMItools (Smith 等,2017)、zUMIs (Parekh 等,2018)、Alevin-fry (He 等,2022)、DropEst (Petukhov 等,2018)、RainDrop (Niebler 等,2020)、baredSC (Lopez-Delisle and Delisle, 2022)、BCseq (Chen and Zheng, 2018) 使用各種質量過濾器和 barcode/UMI 處理策略在一定程度上提高了 CellRanger 的性能。
CellRanger 和 STARsolo 在處理包括 10x Chromium 在內的各種單細胞轉錄組數據集時都具有良好的運行速度,并且準確度極高。但在獲得幾乎相同結果的前提下,后者相比前者提升了至少 5 倍的運行速度,這也驗證了 Alexander Dobin 等人使用 STARsolo 取代 CellRanger 的目的(Brüning 等,2022 ; Chen 等,2021a ; You 等,2021)。
數據處理
在對表達矩陣進行必要的調整(Normalization、Batch Effect Correction、Imputation)后,即可從單細胞轉錄組數據中充分挖掘出生物信息進行分析。Seurat 和 Scanpy 分別基于 R 和 Python 對上述過程進行模塊化、可擴展的處理,是目前單細胞轉錄組數據的主流分析流程(Satija 等,2015 ; Wolf 等,2018)。常規分析流程和預期處理結果可參見總分析框架(圖 4 B-D)。
1、標準化
在測序過程中,技術原因或者細胞本身的生物學差異可能造成同一樣本內(細胞之間)或者不同樣本之間的文庫大小差異(Marinov 等,2014)。無限數方法按照文庫大小進行處理,按照具體原理大致可以分為基于全局縮放的標準化、spike-in 標準化和其他數據變換模型標準化。
全局縮放方法最初是為 bulk RNA 分析而發展起來的,通過特定的縮放因子對全局數據進行縮放(Finak 等,2015)。每萬計數(CPT)變換和每百萬計數(CPM)變換是常見的線性縮放方法,在不考慮 spike-in count 的情況下,它們都對每個 UMI/ 總 UMI count 等距縮放。其他標準化方法包括每百萬 reads 數(RPM)(Mortazavi 等,2008)、修剪均值 M 值(TMM)、DESeq(Robinson and Oshlack, 2010)、上四分位縮放(Bullard 等,2010)、FPKM(Trapnell 等,2010)、RPKM(Tu 等,2012)等,它們對于極值的穩定性比線性縮放更好,因此與 RPKM/FPKM 一樣具有更廣泛的應用范圍。但單獨使用該類方法進行單細胞轉錄組的標準化時,由于數據的稀疏性和假陽性率虛高,效果并不可接受(Evans 等,2018),與特定方法結合時往往需要改進。SCnorm 使用分位數回歸方法來評估不同測序深度依賴細胞組之間的尺度因子(Bacher 等,2017)。bayNorm 基于基因原始計數與真實計數服從負二項(NB)分布的假設,使用集成貝葉斯模型對 scRNA-seq 數據進行標準化(Tang 等,2020)。
spike-in 標準化方法可以看作是全局尺度方法的另一種擴展,因為尺度因子是根據 spike-in 基因計算出來的。需要注意的是,將 RNA spike-ins 的信息添加到其他方法中也可以提高 SCnorm 等標準化的效果。GRM 是一種基于 spike-in ERCC 分子濃度伽馬分布的方法,其中 ERCC 是測序中常用的校準材料(Ding 等,2015)。BASiCS 是一種自動貝葉斯標準化方法,將泊松分層模型應用于 spike-in(技術)基因,以推斷細胞特定的標準化常數(Vallejos 等,2015)。
以上方法都是在細胞內 RNA 數量恒定的假設下對基因進行縮放,而這可能具有欺騙性,因此其他轉化模型采用了不同的策略。由于單細胞轉錄組數據存在零膨脹問題,一些模型就是為此而設計的,例如相對對數表達(RLE)方法 ascend(Senabouth 等,2019)和基于 NB 的模型,如 Dino(Brown 等,2021)、scTransform(Hafemeister and Satija, 2019)。其他轉化模型歸一化方法如 MUREN 使用最小二乘(LTS)回歸算法(Feng and Li, 2021);Sanity 使用從 UMI 計數推斷出的對數轉錄商(LTQ)作為貝葉斯框架的輸入,以避免泊松波動,因為 LTQ 向量的變化估計了基因表達值(Breda 等,2021);PsiNorm 是一種基于無監督帕累托分布尺度參數的方法,用于提升標準化效率和準確率(Borella 等,2021)。Charles Wang 比較了 sctransform、TMM、DESeq 等共 8 種標準化方法,其中 sctransform 和 logCPM(Seurat 的內置處理方法)受數據影響最小,在可變數據集上最穩定(Chen 等,2021a)。
2、批次效應校正
由于實驗設計、測序平臺、測序時間、人員操作流程等原因,不同的單細胞轉錄組測序數據在 mRNA 捕獲效率、測序深度等會存在明顯差異,從而產生樣本間的批次效應(Chen 等,2019a;Hwang 等,2018;Tung 等,2017)。理論上可以通過實驗策略消除技術變異,但由于實驗過程的客觀限制以及測序儀器誤差,不可避免地會或多或少地引入批次效應。利用計算方法進行校正是解決不完善實驗設計的必要手段,通常使用的方法可以分為相互最近鄰(MNN)方法、基于潛在空間的方法、基于圖的方法、DL 方法和其他方法。
MNN 首先識別出不同批次之間同一細胞類型的最相似細胞,然后利用這些細胞進行批次效應校正,包括 batchelor(Haghverdi 等,2018)、Scanorama(Hie 等,2019)、Canek(Loza 等,2022)。另一類使用 MNN 的方法是基于降維后的潛在空間,如 Seurat (Satija 等,2015)、BEER (Zhang 等,2019b)、SMNN (Yang 等,2021a)、iSMNN (Yang 等,2021b)。例如,Seurat 使用典型相關分析 (CCA) 潛在空間中的 MNN 對 (稱為“錨點”)來匹配相似細胞,而 BEER 使用主成分分析 (PCA) 子空間來篩選相似性較差的子群。SMNN 和 iSMNN 分別采用監督機器學習和迭代監督機器學習來細化從預校正細胞聚類或迭代細胞聚類信息中訓練出的 MN 對。
基于潛在空間的方法是指在隱藏空間或降維后的嵌入中進行批次效應校正的方法,除了基于 MNN 聚類的策略外,還有與 PCA 相關的空間方法 Harmony(Korsunsky 等,2019)、FIRM(Ming 等,2022)、Monet(Wagner, 2020);t 分布隨機鄰域嵌入 (tSNE) 空間方法 sc_tSNE(Aliverti 等,2020)和 ZINBWaVE(Gao 等,2019)。Harmony 被廣泛用于去除樣本間的批次效應,使用 PCA 方法將排序的細胞輸入到單個公共嵌入中,然后在最大多樣性聚類和線性批次校正之間迭代循環,直到為每個細胞分配一個特定的校正因子,可用于后續的批次效應去除。Sc_tSNE 方法引入梯度下降算法對傳統 t -SNE 算法進行優化,隨后采用線性校正(Aliverti 等,2021)。ZINB-WaVE 最初設計用于在單細胞數據中進行基因提取,Risso et al.(2018)將該方法擴展至小批量優化。
基于圖的方法利用細胞基因表達矩陣將數字信息轉化為空間構造的圖,其中節點代表不同類型的批次,邊的權重基于不同的計算方法。BBKNN 利用 k 近鄰細胞構建圖(KNN 圖),通過使用均勻流形近似與投影(UMAP)方法合并不同數據集間單個細胞的圖實現批次效應校正,這也是 Scanpy 工作流程中的默認方法(Pola ński 等,2020 ; Wolf 等,2018)。王波在 OCAT 中提出“幽靈細胞”(默認為 k-means 算法聚類中心)來制作細胞連接的二分圖(Wang 等,2022a)。
近年來,深度學習方法的快速發展也為批次效應校正提供了新思路,實現高效、大通量的數據處理,如 INSCT(Simon 等,2021)(三重態神經網絡)、CLEAR(Han 等,2022)(自監督對比學習)、BERMUDA(Wang 等,2019e)(遷移學習)、iMAP(Wang 等,2021a)(VAE-GAN)、ResPAN(Wang 等,2022e)(Wasserstein GAN),一些新方法被證明在批次效應校正方面有更好的效果;例如,基于從 SciBet 學習到的帶注釋數據集中的生物學先驗知識,SSBER 可以在大型 RNA 測序數據集中去除批次效應(Zhang and Wang,2021)。建議在整合單細胞轉錄組數據之前,應根據數據的實際情況先測試多種方法,然后選擇最合適的批次效應去除方法。例如,Jinmiao Chen 團隊和 Charles Wang 團隊分別于 2020 年和 2021 年對本綜述 2.2 中提到的前三種方法的大部分進行了基準測試,證明了 Harmony 和 Seurat V3 在大多數情況下都能達到良好的批次效應校正效果,這符合這兩種方法如今仍然被廣泛使用,但對于深度學習方法來說仍然缺乏好的指標這一事實(Chen 等,2021a;Tran 等,2020)。
3、填補
測序過程中會引入大量 0 值(高通量大規模 10x 基因組測序數據中零值可能超過 90%)(Stegle 等,2015 ; Talwar 等,2018),這會干擾下游生物學差異分析,因此必須對原始基因表達矩陣中的缺失數據值進行填補,同時有效區分技術噪音零值與生物學零值。
基因 / 細胞分離方法主要應用于早期的填補,其分別考慮細胞相似性(MAGIC (van Dijk 等,2018)、Sclmpute (Li and Li, 2018)、VIPER (Chen and Zhou, 2018)、RESCUE (Tracy 等,2019)、scRMD (Chen 等,2020a)、scRoc (Ran 等,2020))或基因間關系(SAVER (Huang 等,2018a)、SAVER-X (Wang 等,2019a)、G253 (Wu 等,2021e)、DCA (Eraslan 等,2019)、DeepImpute (Arisdakessian 等,2019))。總體而言,這些方法缺乏對數據集整體的考慮,容易導致過度插補或者引入誤差(Zhang 等,2019d)。綜合方法綜合考慮細胞與基因之間的聯系:CMF-Impute 和 netNMF-sc 是最早有效利用細胞與基因之間的關聯進行插補的方法(Elyanow 等,2020;Xu 等,2020a)。scIGANs 通過特定的 GAN 模型處理基因表達矩陣,利用生成的細胞訓練 GANs 模型來插補 dropout(Xu 等,2020b)。近年來,新的方法還在不斷被提出,以更好地解決 dropout 之外的技術噪聲對數據的影響,并實現對生物零值的更好的區分。AutoClass(Li 等,2022c)實現了無監督處理,而 ALRA 方法主要針對生物零值(Linderman 等,2022)。scMOO 進行了根本性的改變,利用數據的潛在結構來學習細胞相似性垂直結構和總低秩結構中的深度關聯,從而取得了比單一基因表達矩陣作為輸入更好的插值效果,但對內存的要求也更高(Jin 等,2022a)。sc-PHENIX 利用 PCA-UMAP 初始化方法,實現了基因表達的非線性插值(Padron-Manrique 等,2022),目前哪種插值能取得最佳效果尚無明確定論。由于數據集本身的原因,下游分析的目的會有不同的選擇,但毫無疑問最好的填補方法將能夠以較低的計算要求有效區分技術噪聲零值和生物零值(Jiang 等,2022a;Wen 等,2022)。
4、特征選擇
為了降低數據維數以提升計算分析效率、減少技術噪聲干擾和模型過擬合的風險,我們常常采取特征選擇策略,選取不同細胞中差異較大的基因,而非整個數據集基因進行聚類等后續分析(Brennecke 等,2013;Jackson and Vogel,2022;Svensson 等,2017)。
在 bulk RNA-seq 分析中,尋找差異基因的方法通常包括基于倍數變化(FC)的方法、基于統計檢驗的方法和 FC- 統計檢驗方法,顯然后者的篩選結果和可信度最好(Chung and Storey,2015)。
早期的單細胞特征選擇方法缺乏平均表達量與方差之間的校正,導致結果中高表達基因的比例過高(Brennecke 等,2013)。EDGE 采用大量弱學習器的集成學習方法來學習細胞間相似性概率,提取基于信息熵的顯著貢獻作為高可變基因(Sun 等,2020c)。同樣,SAIC 基于迭代聚類最終輸出實現了最優細胞簇分離(Yang 等,2017)。近期,一些新的特征提取策略被提出并證明了其穩定性和有效性,但它們之間的性能權威驗證尚缺乏:包括基于基因表達分布矩陣的方法 SCMER(Liang 等,2021b)、RgCop(Lall 等,2021)、scPNMF(Song 等,2021a)、SIEVE(Zhang 等,2021e);基于熵的方法 IEntropy(Li 等,2022g)、infohet(Casey 等,2023);綜合考慮聚類的方法有 Triku(Ascensión 等,2022)、FEAST(Su 等,2021)等。由于上述方法絕大多數忽略了整體的依賴于基因表達的特征,因此提出了綜合的方法,如 Triku 使用 k 最近鄰圖的方法對基因表達模式進行綜合探索和分類,實現無偏差地篩選出更有生物學意義的特征基因;FEAST 在共識聚類上通過 f 檢驗對特征進行排序,并基于特征評估算法提取 HVG(Wang 等,2022c)。
其他一些方法使用高可變基因以外的特征來表示數據集,例如 scVEGs 和 scSensitiveGeneDefine 方法,使用高變異系數(CV)作為特征提取;BASiCS 方法利用 spike-in 基因的信息(Chen 等,2016b;Chen 等,2021b)。總體來看,基于準確性、生物學可解釋性等角度,當前特征選擇的主要目標是有效提取 HVG,以便對高維轉錄組數據進行有效的下游分析。
5、降維
由于單細胞轉錄組通常包含數萬個甚至更多的基因,不利于直接提取有效信息,在實際分析過程中,通常需要對原始測序數據進行降維。除了利用前文提到的特征選擇方法處理高維單細胞轉錄組測序數據外,降維也是一種有效的方法,根據降維策略可分為線性降維(基于潛在狄利克雷分配(LDA)的方法、基于 PCA 的方法)和非線性降維(基于 t -SNE 的方法、基于 UMAP 的方法)(Andrews and Hemberg,2018;Becht 等,2019;Laurens and Hinton,2008;Peres-Neto 等,2005)。
在線性降維中,LDA 和 PCA 是兩種廣泛使用的算法,LDA 從分類最大的角度區分特征,而 PCA 則從方差最大的角度正交提取主成分。盡管有 JPCDA、LDA-PLS 等改進算法,但是 LDA 模型在單細胞轉錄組數據中的降維效果仍然不是最優的(Tang 等,2014 ; Zhao 等,2020)。PCA 是另一種線性變換,Seurat 通常根據標準差 -PC 圖的拐點或者 PC 的比例檢驗結果 P 值(ScoreJackStraw 函數)來確定 PC 數量的多少。其他基于 PCA 的降維方法的變體包括 pcaReduce(?urauskien ?和 Yau,2016),GLM-PCA(Townes 等,2019),RPCA(Gogolewski 等,2019),tRPCA(Candès 等,2011),scPCA(Boileau 等,2020),PCAone(Li 等,2022l)。GLM-PCA 將傳統 PCA 分析擴展到非正態分布,通過引入指數家族似然策略直接處理原始矩陣,使 PCA 擺脫正態化限制,然后使用偏差對基因實現進行排序和提取(Collins 等,2002)。ScPCA 使用對比 PCA 和稀疏 PCA 分別去除技術噪音和數據,進一步增加了 PCA 的穩定性(Abid 等,2018 ; Zou 等,2006)。由于大多數 scRNA-seq 數據集難以通過簡單的線性降維進行有效表示,解決這一問題的第一個策略是基于快速 PCA 分析方法。PCAone 提出了一種新的快速隨機奇異值分解(RSVD)策略,在 35 分鐘內完成 130 萬小鼠腦細胞單細胞數據的分析(Li 等,2022l)。
非線性降維是另一種解決方案,如非參數降維方法 t -SNE 和 UMAP,都需要預先設置聚類的超參數;而在分類效果上,前者傾向于離散數據中細胞的形成。在合理使用參數設定的情況下,UMAP 與 t -SNE 并無明顯差異,即在使用相同的信息初始化方法后,二者可以在保留數據集全局結構的同時,產生近似的分析效率(Do and Canzar,2021;Kobak and Linderman,2021)。針對 t -SNE 的改進方法包括 net-SNE、qSNE、FItSNE、聯合 t -SNE(Cho 等,2018a;Linderman 等,2019;Wang 等,2022b),而 UMAP 的改進主要來自于 Leland McInnes 課題組對該方法的自我改進(McInnes 等,2018)。為了更好地可視化 t -SNE 或 UMAP 的降維結果,Hyunghoon Cho 提出了基于局部半徑依賴優化的轉錄組變異信息 den-SNE/densMAP 方法,以迭代優化傳統 t -SNE/UMAP 的功能;Stefan Canzar 提出了 j -SNE/jUMAP 來改善多模態組學數據聯合可視化結果,減少可視化的誤導性(Do and Canzar,2021;Narayan 等,2021)。
6、聚類
在單細胞轉錄組數據分析中,通過聚類將細胞劃分為亞群,從而能夠表征多細胞生物中不同細胞類型,這有助于我們從細胞異質性的角度準確地分析不同的組織或發育過程。聚類的實際效果會受到數據預處理步驟的影響,例如浴效應歸一化、歸納、降維等。
在特征基因選擇和降維之后,絕大多數單細胞是基于距離進行聚類的。K 均值聚類算法的概念被用于 SCUBA、SC3 和 RaceID 等應用(Grün 等,2015;Kiselev 等,2017;Macqueen, 1967;Marco 等,2014)。在參數選擇改進方面,SAIC 通過 Davies-Bouldin 指數迭代優化多個初始中心 K 和 P 值,以獲得最優解;LAK 將參數選擇算法應用于數據集,實現參數的自動選擇(Davies and Bouldin, 1979;Hua 等,2020;Yang 等,2017)。在超高維數據的操作中,LAK 添加 Lasso 懲罰項進行標準化,mbkmeans 使用小批量 k 均值實現百萬細胞級別的快速聚類(Hicks 等,2021)。SMSC 應用譜聚類方法來提高聚類性能,但對于超高維數據會損失一定的準確性(Qi 等,2021)。另一大類廣泛使用的距離聚類方法依賴于共享最近鄰圖結構和圖聚類,其中使用最廣泛的是 Louvain 或 Leiden(Blondel 等,2008;Xu and Su, 2015)。稀有細胞的識別需要結合特定方法進行改進,例如 dropClust 使用局部敏感哈希工作流篩選最近鄰,然后是 Louvain 聚類,它使用指數衰減函數來保留更多稀有細胞的轉錄組特征(Sinha 等,2018)。其他基于距離的聚類方法使用不同的算法核心:SIMLR 使用高斯核學習模型為數據集中潛在的 C 細胞群體構建核矩陣,而 Conos 提出聯合相互最近鄰(mNN)圖聚類來實現對多個不同單細胞轉錄組樣本的整合分析(Barkas 等,2019 ; Wang 等,2017a)。基于密度的聚類利用樣本分布的接近程度進行聚類,DBSCAN 是最經典的算法(Ester 等,1996 ; Fukunaga and Hostetler, 1975)。對于單細胞測序,densityCut 和 FlowGrid 就是基于此原理設計的(Ding 等,2016 ; Fang and Ho, 2021)。層次聚類是一種自下而上的聚類方法,通過無監督學習,不斷重復計算細胞與細胞的相似性進行分類,直至完成預設的聚類數(Guo 等,2015)。隨后,RCA 聚類采用常規的層次聚類方法,對映射到全局參考面板的細胞進行聚類;HGC 在 SNN 圖上構建層次樹(Li 等,2017;Zou 等,2021)。為了解決常規層次聚類方法難以對某一組細胞進行聚類、只允許同一組特征基因進行聚類的缺陷,K2Taxonomer 采用約束 K 均值算法擴展到樣本組,基于多個基因集遞歸進行積分計算,以捕獲各種分辨率下的亞組(“類似分類學的細胞”)(Reed and Monti, 2021)。Mrtree 將層次聚類的策略應用于平面簇的多個劃分,并構造多分辨率協調樹用于細胞聚類(Peng 等,2021a)。最近,Zelig 和 Kaplan(2020)提出了一種 KMD 聚類方法,通過平均鏈接層次聚類模型在聚類時消除了超參數 K,大大減少了主觀性帶來的判斷誤差。
深度學習聚類方法是將機器學習方法與上述單細胞轉錄組聚類策略相結合,可以以無監督、監督或半監督的形式實現更高效的聚類結果。這些方法傾向于學習一種非線性變換,通過將原始高維數據映射到較小的潛在空間中來獲得最佳的低維表示。總體而言,這種方法避免了傳統聚類方法對聚類前數據處理方法選擇的影響。無監督聚類方法包括 ADClust、DESC、SAUCIE、VAE-SNE 等,通常不需要預設聚類個數等參數,以自主學習的方式完成對數據集的分析處理(Amodio 等,2019;Graving and Couzin,2020;Li 等,2020c;Zeng 等,2022c)。雖然無監督聚類方法避免了手動輸入聚類個數等參數,可以延伸到超高維細胞聚類,但有時利用高質量標注數據集或其他先驗知識輔助約束進行監督或半監督聚類,可以實現更為準確的細胞類型分類,提高聚類性能(Bai 等,2021)。基于遷移學習的 ItClust、基于互監督 ZINB 自編碼器和圖神經網絡(GNN)的 scDSC、基于軟 K 均值卷積自編碼器的 ScCAE、基于 Cramer-World 距離最大均值懲罰高斯混合自編碼器的 SeGMA、基于時間序列聚類網絡 STCN 都是廣泛使用的監督聚類(Gan 等,2022 ; Hu 等,2022a ; Hu 等,2020a ; Ma 等,2021b ; Smieja 等,2021)。此外,Zhang 團隊(Yang 等,2023b)利用分層 GAN 設計了另一種廣泛使用的深度學習方法 IMDGC,用于單細胞轉錄組數據分析,以生成的方式構建細胞嵌入簇。
針對聚類中的特殊情況,設計了有針對性的聚類方法:GiniClust(Jiang 等,2016)(更新為 GiniClust 3(Dong and Yuan,2020)、MicroCellClust(Gerniers 等,2021)用于稀有細胞亞群聚類;EDClust(Wei 等,2022)、ENCORE(Song 等,2021b)和 MLG(Lu 等,2021)用于降噪和消除批次效應;ClonoCluster(克隆起源信息)(Richman 等,2023)、IsoCell(可變剪接信息)(Liu 等,2023)使用附加信息進行聚類。Wu 和 Yang 從特征選擇對聚類的影響的角度對聚類方法進行了評估,他們證明更具代表性的特征選擇會提高細胞聚類的水平,基于“聚類相似性”的方法(我們綜述中提到的大多數基于距離的聚類方法)通常具有廣泛的高聚類類型性能;然而,高精度和高運行速度需要根據實際數據集進行有針對性的選擇(Su 等,2021;Yu 等,2022)。雙重浸入 (double dipping) 是一個顯著的問題,即在細胞聚類和差異表達基因中使用相同的表達數據,導致在細胞聚類不正確時 DE 基因的錯誤發現率 (FDR) 過高。例如,如果只存在一個特定的細胞聚類,則不應將任何基因視為差異基因。為了系統地解決這個問題,ClusterDE 采用了聚類對比學習策略進行聚類后 DE 測試。該方法與截斷正態分布 (TN) 檢驗和 Countsplit 方法相比,在不同閾值范圍內具有更好的 FDR 控制 (Song 等,2023a)。
7、細胞類型注釋
細胞分型注釋是指利用特定的信息對單細胞測序數據集中的細胞或細胞亞群進行注釋,作為后續生物學分析的基礎。最常用的策略是對細胞進行無監督聚類,然后根據標記基因進行注釋,例如 scCATCH、SCSA (Cao 等,2020b;Shao 等,2020a),但它難以處理復雜的高維數據集 (Franzén 等,2019;Luecken and Theis, 2019;Zhang 等,2019c)。目前已經開發了多種自動細胞分型方法,大致可分為兩類,即依賴參考和無參考的注釋方法。
依賴參考信息的注釋方法要求用戶提供預先注釋的高質量單細胞轉錄組數據集或來自 PanglaoDB 數據庫、ScType 數據庫等的先驗知識進行比對(Ianevski 等,2022)。根據方法原理的不同,可分為基于層次樹的方法(CHETAH(de Kanter 等,2019)、Garnett(Pliner 等,2019)、HieRFIT(Kaymaz 等,2021)、scHPL(Michielsen 等,2021)、scMRMA(Li 等,2022e)、基于相似性的方法(SingleR(Aran 等,2019)、scmap(Kiselev 等,2018)、deCS(Pei 等,2023)、scID(Boufea 等,2020)、scMatch(Hou 等,2019)、Symphony(Kang 等,2021)、基于簽名基因的方法(Cellassign(Zhang 等,2021)、基于特征基因的方法(Cellassign(Zhang 等,2022)。al., 2019a )、Cell-ID(Cortal 等,2021)、scMAGIC(Zhang 等,2022g)、SciBet(Li 等,2020b)和其他 DL 方法。作為早期方法,ACTINN 是一種使用 3 個隱藏層神經網絡進行注釋分類的深度學習方法(Ma and Pellegrini, 2020)。SCPred 隨后提出了一種基于嵌入的無偏特征選擇的機器學習概率預測方法(Alquicira-Hernandez 等,2019)。其他方法如 Seurat 在 PCA 空間中投影查詢細胞并通過加權投票分類器訓練細胞分型注釋;scSorter 采用高斯混合模型,GraphCS 使用虛擬對抗訓練 (VAT) 損失修改的 GNN 來擴展到多物種、大規模細胞注釋數據集(Guo and Li,2021;Zeng 等,2022a)。
不依賴參考信息的注釋方法使用預先訓練的深度學習模型,可以直接使用查詢數據集作為輸入進行細胞分類。scDeepSort 使用來自人類細胞圖譜 (HCL) 和小鼠細胞圖譜 (MCA) 數據庫的單細胞圖譜作為預訓練加權 GNN 模型的輸入,該模型適用于人類和小鼠細胞注釋并取得良好的效果(Han 等,2018b;Han 等,2020;Shao 等,2021b)。類似地,Pollock 是一個預訓練的人類癌癥參考 VAE 模型,用于對癌癥環境中的多模態細胞進行分類(Storrs 等,2022)。雖然使用起來更方便,但對于差異顯著的查詢數據集難以達到更好的細胞注釋效果,而且由于準確性和預訓練參考數據集的數量也難以擴展應用。還有一些其他用于有針對性領域研究的細胞注釋工具,例如,用于人類腎細胞注釋的 DevKidCC(Wilson 等,2022),用于識別癌癥和正常細胞的 ikarus(Dohmen 等,2021)。總體而言,無參考注釋方法的性能受到預訓練參考數據集的覆蓋率和準確性的制約。
目前,改進細胞注釋工具以在大平臺和多細胞模式下統一分配細胞類型是細胞注釋研究的主流方向,最新的 Cellar 和 ELeFHAnt 方法在這方面做了一些嘗試并取得了初步成果(Hasanaj 等,2022 ; Thorner 等,2021)。總體而言,基于相似性的注釋方法計算量大,在應用于非常大的查詢和參考數據集時,往往會在準確率和速度之間做出權衡,因此一般只適合在較小的數據集中進行細胞分類;對于較大規模的數據集,建議使用 F 檢驗特征選擇或 MLP 分類器(Hu 等,2020a ; Huang and Zhang, 2021 ; Ma 等,2021c)。此外,半監督遷移學習的方法,如 Itclust,在發現新的細胞亞型方面也有不錯的效果。近年來,基于上述參考注釋方法分類的新方法不斷完善,VAE 等深度學習模型也在該領域得到應用。
8、差異表達分析(DEG)
統計檢驗是 Bulk RNA-seq 的差異基因分析中常用到的方法,類似章節 2.4HVG Selection 算法:通常以 P 值和對數倍變化量作為重要參數。統計檢驗包括 t 檢驗(兩個樣本為基礎),Wilcoxon 檢驗,Kolmogorov-Smirnov 檢驗(KS 檢驗),Kruskal-Wallis 檢驗(KW 檢驗),其中一些在單細胞轉錄組 DEGs 的檢驗中也被廣泛使用。基于此,發展了相應的檢測工具:limma(Ritchie 等,2015),edgeR(Robinson 等,2010),DESeq2(Love 等,2014)。limma 和 edgeR 算法均由 Smyth GK 提出,前者基于正態或近似正態分布模型,后者基于過度離散的泊松分布模型。DESeq2 基于 NB 分布模型進行假設檢驗,對 DEG 采用經驗貝葉斯程序。目前 limma 由于特定的分布模型假設,在 RNA 計數分析中誤差較大,雖然 edgeR 和 DESeq2 都利用貝葉斯模型對過度離散進行歸一化,但后者通過數據集 reads 的平均值和異常值檢測促進了 CPM 閾值的篩選,分析效果更好。
單細胞轉錄組 DEG 按照時間和分析方法大致可以分為早期零值參數檢驗、非參數檢驗和其他方法。由于 scRNA-seq 數據中存在大量零數,早期的方法大多基于此觀察做參數檢驗,例如 Monocle (Trapnell 等,2014)、SCDE (Kharchenko 等,2014)、MAST (Finak 等,2015)、scDD (Korthauer 等,2016)、D3E (Delmans and Hemberg, 2016)、TASC (Jia 等,2017)、DEsingle (Miao 等,2018) 和 HIPPO (Kim 等,2020b)。對以上一些方法的評測表明,雖然它們在單細胞數據集的分析中普遍取得了不錯的效果,但對于批量數據(Soneson and Robinson, 2018)相比 DEA 方法并沒有明顯的性能提升。對于不同的數據集,有可能沒有最好的分布模型,因此一種替代解決方案是考慮非參數 DEA 方法。
非參數檢驗或無分布檢驗不需要對數據分布形式做事先假設,因此適用于多數據集的分析,常用方法有 Swish(Zhu 等,2019a)、IDEAS(Zhang 等,2022d)、ccdf(Gauthier 等,2021)、distinct(Tiberi 等,2022)。Swish 通過 Salmon Gibbs 評估轉錄本水平,然后用 Mann-Whitney Wilcoxon 檢驗計算 FC 值。IDEAS 是一種使用 Jensen-Shannon 散度(JSD)或 Wasserstein 距離(Was)進行基因差異表達測量的偽 F 統計量檢驗,P 值由基于 PERMANOVA 的距離測試器基于核的回歸生成。Ccdf 是一種依賴條件累積分布函數的條件獨立性檢驗,通過多元回歸模型預測 DEG。Distinct 提出了一種分層非參數置換方法,使用經驗累積分布函數 (ECDF) 的總距離進行 DEG 識別。替代方法包括深度學習策略 MRFscRNAseq (Li 等,2021a)、基于擬時序推斷的 PseudotimeDE (Song and Li, 2021)、基于非預聚類的 singleCellHaystack (Vandenbon and Diez, 2020)、基于多重評分的 MarcoPolo (Kim 等,2022)。建議不同的單細胞轉錄組數據集應采用數據特定的 DEGs 檢測策略,以優化 DEGs 分析,基于 scCODE 工作流程,可以使用涉及 CDO(DE 基因順序)和 AUCC(一致性曲線下面積)的指標找到最優化的 DEGs 方法(Zou 等,2022)。此外,研究方法在不同的研究背景下會有特定的研究取向,例如在給藥后的劑量反應研究中,DEGs 分析、LRT 線性檢驗和貝葉斯多組檢驗均比其他方法有更好的結果(Nault 等,2022)。
9、可視化
單細胞轉錄組數據分析可視化是指將上述分析結果以圖形的形式直觀地呈現,ggplot2 是 R 中最廣泛的可視化工具,在 R 中被廣泛使用,可以大大增強繪圖能力(Wickham,2009)。ARL 是另一個專門顯示標記基因關聯圖并可顯示其在每個簇中的特征的 R 包(Gralinska 等,2022)。此外,還有其他專門用于標記基因可視化的包,如 Complex Heatmap,本文不再詳細介紹。HVG 可視化通常以火山圖的形式呈現,默認情況下,圖的左側和右側部分分別是代表性不足的基因和代表性過高的基因,而中間是恒定基因。Enhanced Volcano 是一個專門用于繪制火山圖的 R 包,默認情況下也可以使用 ggplot2 來獲得更好的結果。簇可視化通常以 PCA 圖、t-SNE 圖和 UMAP 圖呈現,但值得注意的是,可視化的結果非常具有欺騙性,因為一些小的細胞亞群可能代表 UMAP 圖中顯示的大量細胞。為了解決這些問題,提出了 den-SNE/densMAP、j-SNE/j-UMAP 等改進方法(Macqueen,1967;Marco 等,2014)。此外,FastProject 可以輸出注釋簇的 2D 顯示,PieParty 可以在簇 2D 圖中為每個基因繪制顏色圖(DeTomaso and Yosef,2016;Kurtenbach 等,2021)。
同時,單細胞轉錄組數據的交互式可視化是目前的熱門領域,諸如 Single Cell Explorer 等軟件可以一定程度上實現交互式可視化,但仍需增加交互自由度,以提供更全面的單細胞轉錄組數據 3D 呈現(Cakir 等,2020;Feng 等,2019)。為此,CellexalVR 利用 VR 理論進行交互可視化;CellView 是一個基于 Web 的工具,包括用于不同用途的探索選項卡、共表達選項卡、子簇分析選項卡模塊;Cellxgene VIP 是一個基于 cellxgene 框架的插件,并擴展到基于多個模塊組合的 ST 數據的交互式可視化(Bolisetty 等,2017 ; Legetth 等,2021 ; Li 等,2022f)。
10、單細胞模擬
隨著單細胞轉錄組方法的不斷擴展,基準測試成為了重要挑戰,關鍵問題是需要穩定可靠的數據,因為單細胞轉錄組的直接測序可能缺乏基本事實。真實的單細胞模擬數據為基準測試提供了已知的事實,允許使用真實數據進行訓練,同時匹配實際數據的特征。此外,模擬數據比真實數據提供了更大的靈活性,使分析師能夠根據特定的測試方法調整諸如 dropout rate 等參數。
Splatter 是一個兩步模擬框架,首先模擬來自真實數據的估計參數,然后合并來自用戶的額外參數(Zappia 等,2017)。其六個預先設計的管道模塊接口確保了數據生成的可重復性。最近的更新側重于專業化和泛化。在專業化領域,splaPop 生成具有遺傳效應(數量性狀基因座)的人口規模數據,而 dyngen 模擬動態細胞過程,如發育軌跡(Azodi 等,2021;Cannoodt 等,2021)。在泛化領域,Li 的團隊介紹了理想模擬的六個概念,包括真實性、基因的保存、基因相關性的捕獲、穩健性、參數可調性和效率(Song 等,2023b;Sun 等,2021)。隨后,scDesign2 提出來滿足所有 6 個屬性(Sun 等,2021),接著是 scDesign3,解決單細胞組學統計模擬的空白(Song 等,2023b)。模擬準確性和透明度的提高增強了不同單細胞數據處理方法之間的基準測試,指導選擇最合適的方法以滿足特定數據和許可需求。
