 |  |

基因表達(dá)具有時間和空間的特異性,通過對不同時間點的樣本取材,使用單細(xì)胞轉(zhuǎn)錄組測序技術(shù)能夠解析時間維度上細(xì)胞類型和基因表達(dá)的變化過程。然而單細(xì)胞測序?qū)嶒灥那疤崾墙M織必須通過機(jī)械分離或酶解消化成單細(xì)胞懸液,此過程不可避免的丟失了組織中細(xì)胞所處的原始位置信息,也導(dǎo)致了細(xì)胞間的通訊網(wǎng)絡(luò)被打破,這使我們難以獲得組織中不同區(qū)域的細(xì)胞構(gòu)成和基因表達(dá)狀態(tài),以及不同功能區(qū)之間的基因差異表達(dá)等信息。現(xiàn)有的原位表達(dá)圖譜主要是通過報告基因或原位雜交等技術(shù)來實現(xiàn),但是這些方法實現(xiàn)比較困難,并且通量低,限制了多樣本、高時效的應(yīng)用。而空間轉(zhuǎn)錄組技術(shù)則可以高 效的檢測組織中空間原始位置上的基因表達(dá)模式。
▲圖 空間轉(zhuǎn)錄組被評為 2020 年度技術(shù)
注:圖片來源互聯(lián)網(wǎng) - 侵刪
空間轉(zhuǎn)錄組(Spatial Transcriptomics)就是將基因的表達(dá)情況與關(guān)注的組織切片的免疫化學(xué)染色圖像進(jìn)行整合,從而將組織內(nèi)不同細(xì)胞的基因表達(dá)信息定位到組織的原始空間位置上去,進(jìn)而直接觀測組織中不同部位功能區(qū)基因表達(dá)的差異。空間轉(zhuǎn)錄組技術(shù)利用了常規(guī)的原位技術(shù)和組學(xué)技術(shù)兩方面的優(yōu)勢。實際上空間轉(zhuǎn)錄組已不是新名詞,2016 年 Joakim Lundeberg 的 Spatial Transcriptomics 技術(shù)在 Science 上發(fā)表,2017 年景乃禾老師的 GEO-seq 技術(shù)在 Nature Protocols 上發(fā)表 [2]。目前已發(fā)表的關(guān)于空間轉(zhuǎn)錄組技術(shù)有主要有 Spatial Transcriptomics, Slide-seq, LCM-seq, seqFISH, MERFISH, Liver single cell zonation, Geo-seq 和 Tomo-seq, 涉及物種有人、小鼠、果蠅、秀麗隱桿線蟲、斑馬魚、擬南芥、楊樹和云杉等。
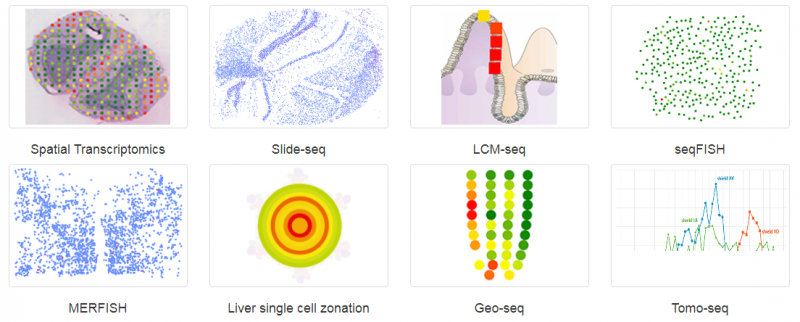
▲圖 常見空間轉(zhuǎn)錄組技術(shù)
注:圖片來源互聯(lián)網(wǎng) - 侵刪
其中,瑞典皇家理工學(xué)院的 Joakim Lundeberg,基于芯片和空間條形碼技術(shù)發(fā)明了高通量的空間轉(zhuǎn)錄組測序方法,并創(chuàng)建了 Spatial Transcriptomics 公司。2018 年底 10X Genomics 宣布收購 Spatial Transcriptomics,并于 2019 年發(fā)布 Visium 空間基因表達(dá)解決方案(Visium Spatial Gene Expression Solution)。10X Genomics 公司提供的 Visium 空間基因表達(dá)解決方案是高通量空間轉(zhuǎn)錄組的商業(yè)化解決方案,其可以檢測完整組織切片的總 mRNA,將總 mRNA 的空間信息與形態(tài)學(xué)內(nèi)容相結(jié)合,并繪制所有基因表達(dá)發(fā)生的位置,獲得完整的基因表達(dá)圖譜;在確定不同細(xì)胞群的同時保留空間位置,為細(xì)胞功能、表型和組織微環(huán)境中位置的關(guān)系提供了重要信息。

空間轉(zhuǎn)錄組學(xué)技術(shù)優(yōu)勢:是轉(zhuǎn)錄組學(xué)研究領(lǐng)域的新方向,也是研究細(xì)胞異質(zhì)性方面的新方法。
(1)準(zhǔn)確定位:探針有效定位組織中 RNA 空間位置,實時了解組織中的轉(zhuǎn)錄組天然狀態(tài)。
(2)簡單易行:芯片設(shè)計簡單易操作、重復(fù)性高、可快速獲得高分辨率空間轉(zhuǎn)錄組信息。
(3)適用性廣:有效應(yīng)用于發(fā)育生物學(xué)、腫瘤生物學(xué)、腦神經(jīng)科學(xué)、植物研究等各生物學(xué)領(lǐng)域。
1、伯豪個性化方案:空間轉(zhuǎn)錄組測序研究需求千差萬別,伯豪生物專業(yè)科研團(tuán)隊針對客戶需求一對一項目建議,為客戶定制空間基因表達(dá)解決方案。
2、組織保存液:空間轉(zhuǎn)錄組技術(shù)對樣本質(zhì)量要求高,新鮮組織樣本需要立即冷凍包埋才能更好的保持樣本 RNA 質(zhì)量,從而保障實驗準(zhǔn)確性。伯豪生物自研了伯優(yōu)?組織保存液,4℃條件下新鮮組織樣本離體 48 小時,細(xì)胞活性及細(xì)胞形態(tài)結(jié)果無明顯影響,有效解決樣本采集→保存運輸→冷凍包埋的不良影響。
3、高質(zhì)量組織切片制備:伯豪生物科研服務(wù)團(tuán)隊經(jīng)大量項目樣本經(jīng)驗累積,探索出 對多種類型組織制備出高質(zhì)量切片方法:“不同組織類型的切片需要優(yōu)化不同的條件”。
4、全面的生信分析流程和個性化數(shù)據(jù)分析。

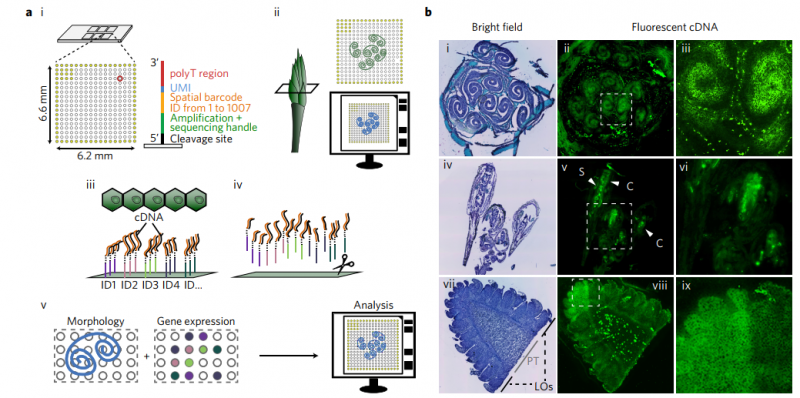
將冷凍組織切片放置在空間轉(zhuǎn)錄組芯片的捕獲區(qū)域內(nèi),進(jìn)行 HE 染色和成像后,對組織切片進(jìn)行透化處理,細(xì)胞內(nèi)的 mRNA 釋放,從而被芯片上帶有 oligo-dT 的探針捕獲,并且每個探針都帶有特異的位置信息(Spatial barcode),然后以 mRNA 為模版進(jìn)行 cDNA 合成,構(gòu)建文庫后再通過測序,獲得基因表達(dá)信息的同時,每一條測序 reads 因帶有 Spatial barcode,從而能夠獲得基因表達(dá)的位置信息。
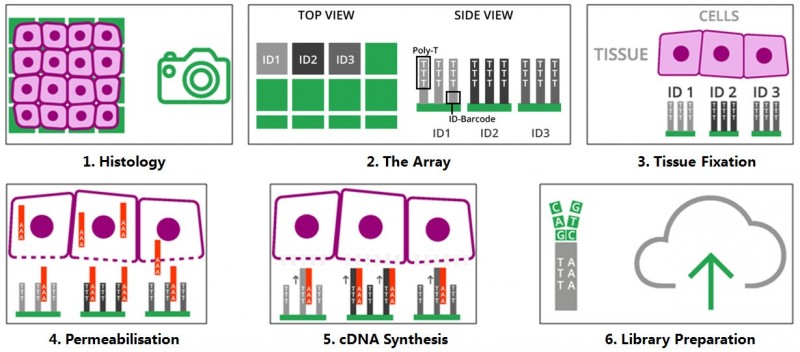
▲圖 空間轉(zhuǎn)錄組技術(shù)的原理
注:圖片來源互聯(lián)網(wǎng) - 侵刪
空間轉(zhuǎn)錄組技術(shù)包含兩種芯片,分別為組織優(yōu)化芯片(Tissue Optimization)和基因表達(dá)芯片(Library Preparation)。組織優(yōu)化芯片用來摸索組織透化的時間,基因表達(dá)芯片用來進(jìn)行正式樣本的空間轉(zhuǎn)錄組實驗。其中基因表達(dá)芯片上有 4 個捕獲區(qū)域,每個區(qū)域大小為 6.5mm×6.5mm,每個捕獲區(qū)域中有 5000 個帶有特異地址序列的探針簇,稱為 barcoded spots,每個 spot 直徑為 55um,包含數(shù)百萬個用于捕獲的 oligo 探針序列,相鄰兩個 spot 的中心距離為 100um。探針序列的結(jié)構(gòu)為:測序引物結(jié)合序列,16nt 的位置序列,12nt 的 UMI 序列以及 30nt 的 oligo-dT 序列。

▲圖 空間轉(zhuǎn)錄組兩種芯片
注:圖片來源互聯(lián)網(wǎng) - 侵刪
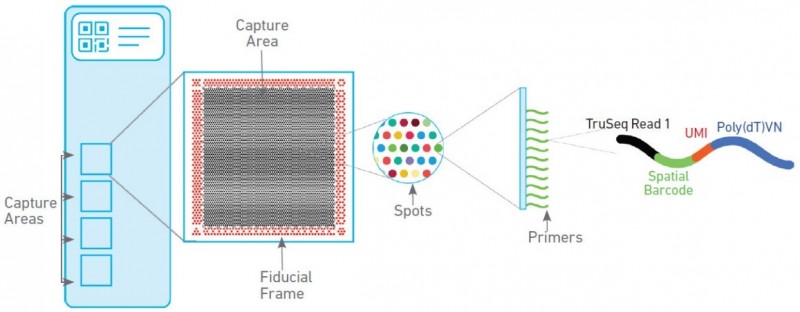
▲圖 空間轉(zhuǎn)錄組基因表達(dá)芯片工作原理
注:圖片來源互聯(lián)網(wǎng) - 侵刪

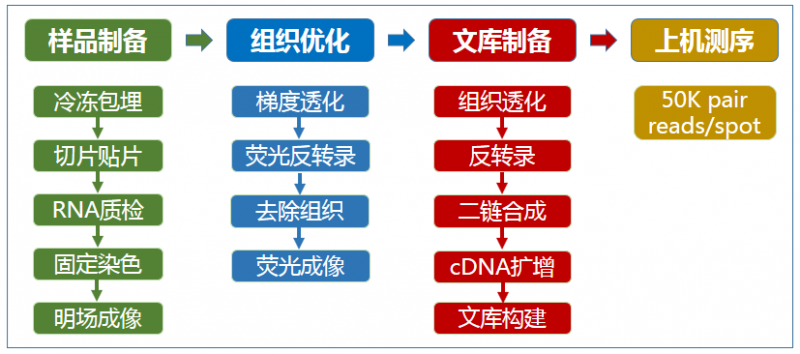
▲圖 空間轉(zhuǎn)錄組測序?qū)嶒灹鞒?

方法一:組織冷凍(不常用)
1、異戊烷和液氮浴:如圖所示,不銹鋼燒杯內(nèi)倒入異戊烷至 2 / 3 體積,然后將不銹鋼燒杯放入液氮中(與異戊烷液位相同),孵育 15 分鐘。
2、新鮮樣本可用 PBS 沖洗,去除殘留血液,然后使用實驗室紙巾吸干組織表面多余的血液或液體,防止冰晶的形成(注:組織長和寬不可超過 6mm±0.2mm,否則后續(xù)實驗無法貼片)。
3、用鑷子或刮刀將組織整體浸沒在異戊烷中,直至整體冰凍(注:冷凍時間可根據(jù)組織類型和大小而改變)。
4、冷凍后,取出組織轉(zhuǎn)移到預(yù)冷的密封容器中,干冰轉(zhuǎn)移至 -80°長期保存或立即進(jìn)行下一步(注:為防止組織樣品蒸發(fā)和脫水,冷凍的組織樣品必須儲存在密封容器中以長期保存)。
方法二:冷凍組織包埋(常用)
1、粉狀干冰:用研缽和杵準(zhǔn)備干冰粉。
2、冷凍 OCT:將 OCT 放在冰上≥30 分鐘(注:OCT 請使用指定品牌)。
3、預(yù)冷鑷子:將鑷子放在干冰中≥30 分鐘。
4、包埋盒上需標(biāo)記組織樣本的方向(注:在添加 OCT 和組織之前,需先在包埋盒上標(biāo)記,因為一旦凍結(jié),OCT 將迅速變成白色,這使得以后很難確定組織方向)。
5、用冷卻的 OCT 鋪平包埋盒底部,避免產(chǎn)生氣泡。
6、從不銹鋼燒杯中取出冰凍的組織,加入到 OCT 的包埋盒中心位置,繼續(xù)倒入 OCT 覆蓋樣本。需避免氣泡產(chǎn)生,尤其在組織附近。
7、立即將含有組織和 OCT 的包埋盒放在干冰粉上,直至整體凍結(jié)(約 20min 以上)。
8、將包埋盒放置到密封袋中,干冰運輸。
注意事項
1、新鮮組織樣本包埋
目前新鮮組織的包埋方法有兩種,一種是液氮 + 異戊烷法;另一種的干冰法。對于臨床手術(shù)切下來的組織樣本一般使用干冰的包埋方法。對于穿刺樣本等一些較小,較輕的樣本,一般推薦用液氮 + 異戊烷的方法進(jìn)行冷凍。OCT 包埋組織塊可以在–80oC 的密封容器中長期保存,或立即進(jìn)行冷凍切片。
2、冷凍切片切、組織樣本質(zhì)控及貼片
由于空間轉(zhuǎn)錄組檢測的是組織中的 RNA,因此要對切片中的 RNA 質(zhì)量進(jìn)行檢測。我們一般取 10 片組織切片進(jìn)行 RNA 抽提并質(zhì)檢,確定組織中 RNA 完整性(RIN>7)。所以要求我們的組織樣本要保證可以切到至少 20 片 10um 厚度的切片,以便完成所有的實驗。
3、組織優(yōu)化
組織優(yōu)化的目的是摸索樣本的透化條件,保證組織切片中的 mRNA 能夠充分釋放。該步驟是獲取真實實驗結(jié)果的必要條件。否則,我們無法判斷是基因表達(dá)高低到底是因為透化不充分導(dǎo)致的,還是實際就是這個樣子。因此:每個樣本建議都要做透化,尤其是臨床樣本。
4、成像
應(yīng)使用 Visium Imaging Test Slide 驗證成像設(shè)置。明場成像基準(zhǔn)框和基準(zhǔn)標(biāo)記應(yīng)清晰可見,并使用 Brightfeld 設(shè)置聚焦。Visium Imaging Test Slide 四個區(qū)域(A1,B2,C1,D2)具有熒光斑點,可通過 TRITC 和 Cy5 flter cubes 檢測到,熒光設(shè)置應(yīng)清晰可見 A1,B2,C1 和 D2 中的熒光點,且熒光點信號應(yīng)從左到右減小。
5、正式實驗
正式實驗時要對反轉(zhuǎn)錄后的 cDNA 長度分布,濃度和量進(jìn)行判斷。cDNA 的長度分布在~200bp-9000bp 之間,在 1000bp 左右會有峰值(不同的組織類型會有些許差異)。
6、測序
在捕獲區(qū)域,每個組織覆蓋的 spot 建議至少測 50000 read pairs。整個捕獲區(qū)域共有 5000 個 spots。可以根據(jù)組織貼到芯片上后,覆蓋芯片的大小來判斷測序的數(shù)據(jù)量。
計算公式(Coverage Area x total spots on the Capture Area)x 50,000 read pairs/spot 例如:組織覆蓋了 60% 的區(qū)域,則數(shù)據(jù)量為(0.60 x 5,000 total spots) x 50,000 read pairs/spot=150 million read pairs。

圖 切片覆蓋芯片區(qū)域百分比

空間轉(zhuǎn)錄組數(shù)據(jù)分析的核心是根據(jù)每個芯片上每個 spot 的基因表達(dá)信息進(jìn)行聚類,然后將 spot 根據(jù)坐標(biāo)位置序列放回到組織的圖像上,同時可以對每個 gene 在組織上表達(dá)的空間位置進(jìn)行定位。
獲得測序數(shù)據(jù)后,首先利用 Space Ranger 軟件可以自動化的對圖像進(jìn)行處理、數(shù)據(jù)比對和 Barcode 處理。另外一個軟件 Loupe Cell Browser 是一個適用于 Windows 和 MacOS 的桌面應(yīng)用程序,它可以快速、輕松地可視化和分析 10X Visium 數(shù)據(jù)。伯豪生物除了提供 spot 基因數(shù)和 UMI 數(shù)統(tǒng)計、切片 spot 聚類和聚類亞群 marker 基因分析等基礎(chǔ)和高級分外,同時還提供個性化分析,如特定 pathway 功能富集分析等。
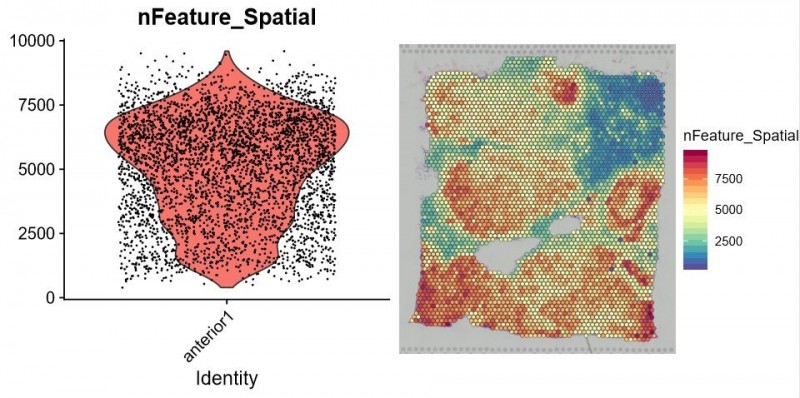
▲圖 每個 spot 特異表達(dá)的基因數(shù)統(tǒng)計
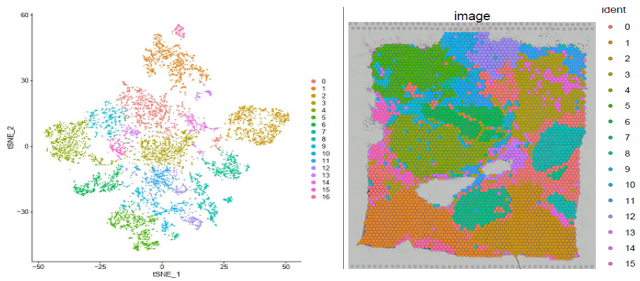
▲圖 聚類結(jié)果及切片 spot 位置分布展示
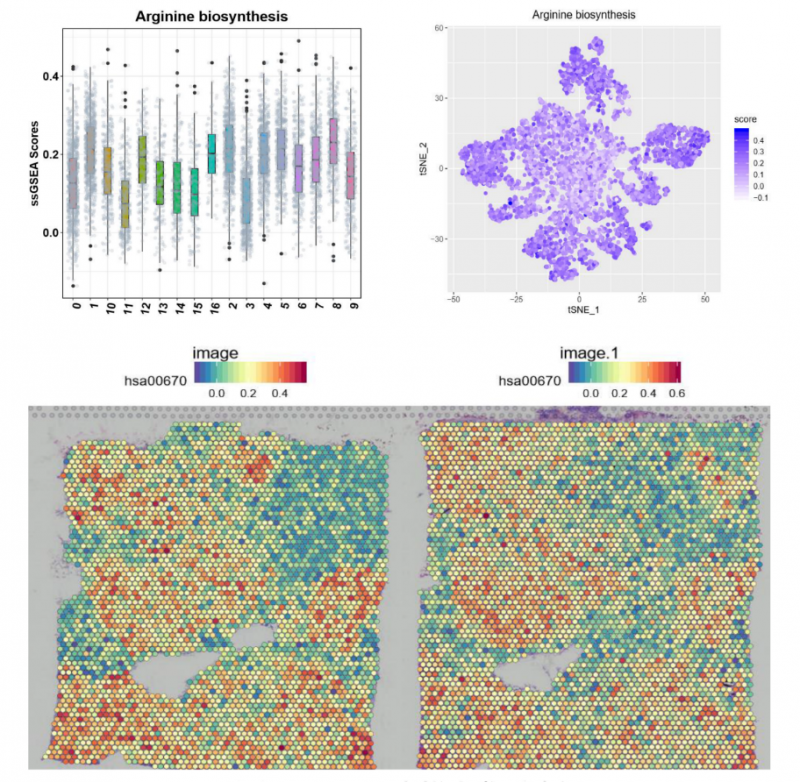
▲圖 特定 pathway 功能富集分析
結(jié)合組織區(qū)域分布對數(shù)據(jù)進(jìn)行挖掘
大部分組織其實是有其特定的區(qū)域劃分的,比如說大腦里有皮層、丘腦、海馬、脈絡(luò)叢等多個區(qū)域。將組織的區(qū)域劃分和亞群(或細(xì)胞類型)的分布結(jié)合起來還是能發(fā)現(xiàn)很多有價值的信息的。
可以根據(jù)不同區(qū)域特異表達(dá)的 maker 基因的分布來判斷每個區(qū)域在組織切片上的位置。例如皮層 marker 基因 STX1A 的表達(dá)分布,海馬 marker 基因 HPCA 的表達(dá)分布等。
結(jié)合病理學(xué)特征對數(shù)據(jù)進(jìn)行挖掘
空間轉(zhuǎn)錄組技術(shù)正真的精髓不是研究細(xì)胞亞群的分布,而在于將它在空間位置上體現(xiàn)的異質(zhì)性跟組織病理學(xué)特征的分布進(jìn)行結(jié)合,挖掘在不同病理學(xué)特征下轉(zhuǎn)錄組學(xué)的差異。這對于研究疾病病變的機(jī)制、幫助臨床實現(xiàn)更好的患者分子分型、以及空間位置 Biomarker 的挖掘方面都是非常有價值的。通過手動把這些區(qū)域圈出來進(jìn)行轉(zhuǎn)錄組層面的比較,找出不同病灶區(qū)的特異性 marker,分析疾病在一步步發(fā)展進(jìn)程中生物學(xué)功能的變化,甚至可以思考一下是否能找出一些關(guān)鍵性因子來阻斷疾病的進(jìn)展。
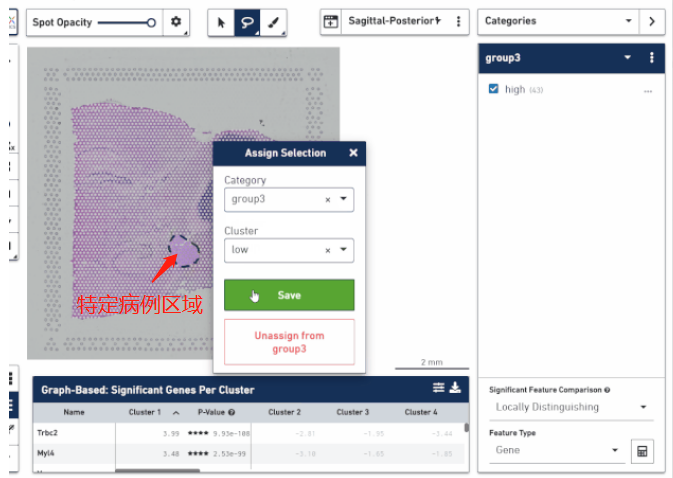
▲圖 根據(jù)病理信息選取特定區(qū)域分析
空間轉(zhuǎn)錄組聯(lián)合單細(xì)胞 RNA 測序解析細(xì)胞類型的空間位置信息(Multimodal intersection analysis,MIA)
空間轉(zhuǎn)錄組測序可以獲得不同基因在組織切片上的空間位置信息,但不能獲得詳細(xì)的細(xì)胞類群信息(空間轉(zhuǎn)錄組不是單細(xì)胞分辨率,只能粗略的分析切片上不同位置的細(xì)胞類型)。因此,需要借助但細(xì)胞測序數(shù)據(jù)來分析細(xì)胞類型,然后通過生物信息學(xué)的分析方法將單細(xì)胞類群映射到空間轉(zhuǎn)錄組數(shù)據(jù)上。
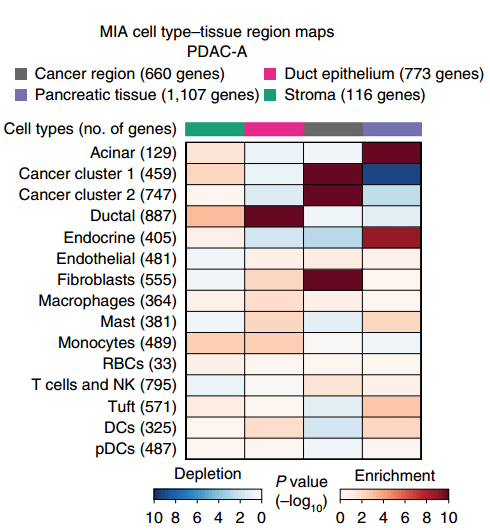
▲圖 MIA 熱圖
備注:MIA 熱圖,上方的顏色條反映了 ST 區(qū)域的子聚類(cancer region,Pancreatic tissue,Duct epithelium 和 stroma)。左側(cè)代表不同的細(xì)胞類群。色塊代表 enrichment 或者 depletion。Enrichment 代表該細(xì)胞類群富集到了該區(qū)域。Depletion 代表該細(xì)胞類群在該區(qū)域缺失。

空間位置信息,或者細(xì)胞在組織中天然的狀態(tài)在研究過程中其實具有十分重要的價值,特別針對某些研究領(lǐng)域,如發(fā)育生物學(xué)(不同位置的細(xì)胞接受不同的信號濃度梯度、響應(yīng)不同的外界刺激,具有不同的發(fā)育命運)、腫瘤生物學(xué)(腫瘤組織與癌旁組織的區(qū)別,腫瘤細(xì)胞侵潤過程中腫瘤細(xì)胞的變化與對正常細(xì)胞的影響,腫瘤轉(zhuǎn)移的不同過程階段等)、腦神經(jīng)科學(xué)(不同腦區(qū)位置的神經(jīng)元結(jié)構(gòu)、神經(jīng)連結(jié),中間神經(jīng)元投射,突觸前后,神經(jīng)膠質(zhì)相互影響等等),細(xì)胞來源的位置信息是極為關(guān)鍵的決定因素。
空間轉(zhuǎn)錄組測序技術(shù)除了在人和動物上得到了廣泛的應(yīng)用之外,在植物領(lǐng)域也有所突破。2017 年發(fā)表在 nature plant 上的一篇文章就闡釋了空間轉(zhuǎn)錄組在擬南芥中的應(yīng)用,利用空間數(shù)據(jù)作者分析了 141 個基因的表達(dá)水平差異,8 個花序組織域中 189 條通路。伯豪生物作為較早的空間轉(zhuǎn)錄組測序技術(shù)服務(wù)商,也在植物的空間轉(zhuǎn)庫組領(lǐng)域有所突破。開發(fā)了多種植物組織的空間轉(zhuǎn)錄組樣本制備,透化條件摸索等。獲得了寶貴的項目經(jīng)驗。
常見空間轉(zhuǎn)錄組的應(yīng)用方向主要在腫瘤學(xué),免疫學(xué),發(fā)育生物學(xué),神經(jīng)科學(xué)及病理學(xué)等方向。
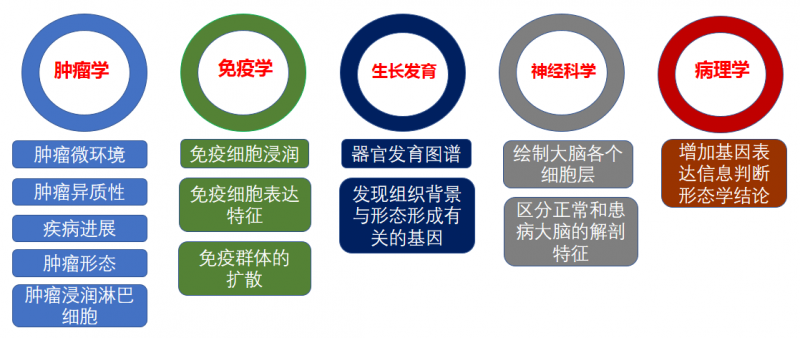
▲圖 空間轉(zhuǎn)錄組的應(yīng)用方向
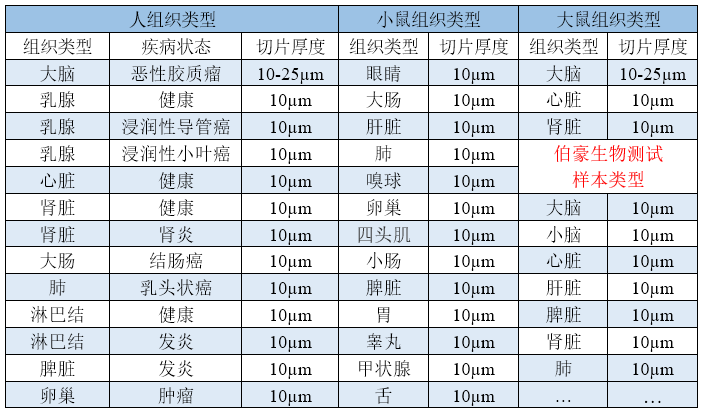
▲表 已做過優(yōu)化的樣本類型

案例一
空間轉(zhuǎn)錄組學(xué)聯(lián)合單細(xì)胞 RNA-seq 揭示胰腺導(dǎo)管腺癌的組織結(jié)構(gòu)
Integrating microarray-based spatial transcriptomics and single-cell RNA-seq
reveals tissue architecture in pancreatic ductal adenocarcinomas [5]
發(fā)表雜志:Nature biotechnology
影響因子:36.558
發(fā)表時間:2020 年 1 月
單細(xì)胞 RNA 測序(single RNA sequence,scRNA-seq) 能夠系統(tǒng)地識別組織中的細(xì)胞類群,但它不能獲取各個細(xì)胞類型在組織中的空間位置信息。空間轉(zhuǎn)錄組測序(spatial transcriptomics,ST),可以獲得不同基因在組織切片上的空間位置信息,但不能獲得詳細(xì)的細(xì)胞類群信息。作者通過對胰腺導(dǎo)管癌病人的同一樣本同時進(jìn)行 scRNA 測序和 ST 測序。同時開發(fā)了一種多模態(tài)交叉分析方法(Multimodal intersection analysis,MIA)來將單細(xì)胞測序數(shù)據(jù)映射到空間轉(zhuǎn)錄組數(shù)據(jù)上,獲得各種細(xì)胞類型在組織上的空間分布。研究發(fā)現(xiàn)導(dǎo)管細(xì)胞、巨噬細(xì)胞、樹突狀細(xì)胞和癌細(xì)胞的亞群在不同的空間位置區(qū)域上富集或丟失,以及與其他類型的細(xì)胞有顯著的共富集。此外,作者還發(fā)現(xiàn)了炎癥成纖維細(xì)胞和表達(dá)應(yīng)激反應(yīng)基因模塊的癌細(xì)胞的定位。
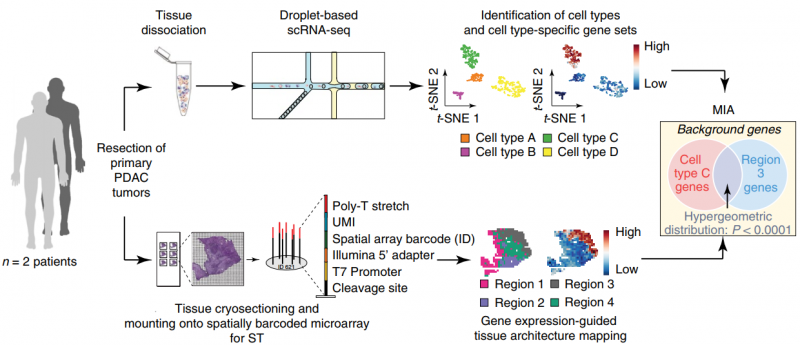
圖:ST 聯(lián)合 scRNA 揭示胰腺導(dǎo)管癌組織細(xì)胞類型及結(jié)構(gòu)
案例二
空間轉(zhuǎn)錄組聯(lián)合單細(xì)胞測序揭示心臟發(fā)育
A Spatiotemporal Organ-Wide Gene Expression and Cell Atlas of the Developing Human Heart[11]
發(fā)表雜志:Cell
影響因子:38.637
發(fā)表時間:2019 年 12 月
摘要:人類心臟形態(tài)發(fā)生的過程尚不清楚。它的完整特性需要用單細(xì)胞空間分辨率深入探索器官范圍內(nèi)基因表達(dá)的協(xié)調(diào)。在這里,我們提出了一種分子方法,揭示了在三個發(fā)育階段的胚胎心臟細(xì)胞類型的轉(zhuǎn)錄景觀,并將細(xì)胞類型特異性基因表達(dá)映射到特定的解剖結(jié)構(gòu)域。空間轉(zhuǎn)錄組學(xué)鑒定了在每個發(fā)育階段對應(yīng)不同解剖區(qū)域的獨特基因譜。作者通過對人類心臟發(fā)育有三個階段:孕早期 4.5- 5 周,6.5 周和懷孕后 9 周的人類胚胎心臟樣品進(jìn)行單細(xì)胞 RNA 測序鑒定的人類胚胎心臟細(xì)胞類型,同時通過空間轉(zhuǎn)錄組測序技術(shù)來獲取基因表達(dá)的空間位置信息。然后使用原位測序來細(xì)化這些結(jié)果,并為三個發(fā)育階段創(chuàng)建一個空間亞細(xì)胞圖譜。形成了一個公開的人類心臟發(fā)育的網(wǎng)絡(luò)資源,以促進(jìn)未來對人類心臟發(fā)生的研究。
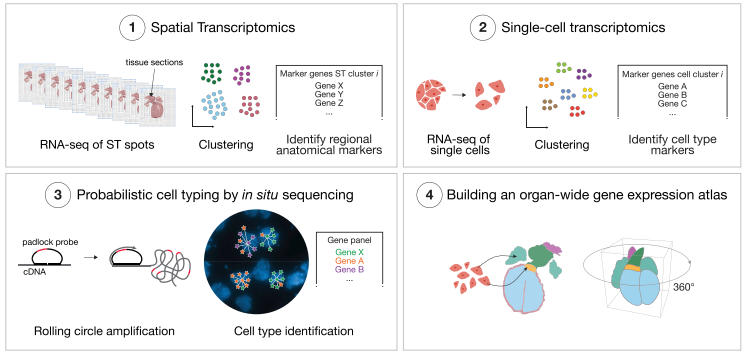
圖 18 實驗設(shè)計思路
A、本研究包括三種人類心臟組織的發(fā)育階段。B、本研究所采用的分子生物學(xué)方法:(1)利用空間轉(zhuǎn)錄組技術(shù)(ST),鑒定出了解剖學(xué)區(qū)域特異的 marker;(2)用 scRNA-seq 技術(shù)分析心臟中間時間點的細(xì)胞類型異質(zhì)性;(3)用 ISS 技術(shù)定位亞細(xì)胞分辨率的關(guān)鍵基因;(4)構(gòu)建器官水平的 3D 基因表達(dá)圖譜。
案例三
單細(xì)胞 RNA 測序 & 空間轉(zhuǎn)錄組測序“強(qiáng)強(qiáng)聯(lián)手”揭示腸道發(fā)育
Spatiotemporal analysis of human intestinal development at single-cell resolution
發(fā)表期刊:Cell
影響因子:38.637
發(fā)表時間:2021 年 1 月
腸道是人體大的屏障器官,與腸道微生物共生協(xié)調(diào)營養(yǎng)需求和免疫。多種相互關(guān)聯(lián)的細(xì)胞類型構(gòu)成了成熟的腸道及其不同的形態(tài),但其發(fā)育的分子基礎(chǔ)仍不清楚。來自牛津大學(xué) Simmons 和 Koohy 教授團(tuán)隊通過單細(xì)胞測序聯(lián)合空間轉(zhuǎn)錄組技術(shù)來研究腸道發(fā)育過程中形態(tài)的變化。研究對來自 17 例胚胎的 77 個樣本進(jìn)行了單細(xì)胞 RNA 測序(使用寡核苷酸標(biāo)記抗體的多重混樣技術(shù))以及對來自 5 個樣本的 8 張切片進(jìn)行了空間轉(zhuǎn)錄組測序。通過一系列的生物信息學(xué)分析方法,包括細(xì)胞間通訊網(wǎng)絡(luò)構(gòu)建,SCENIC 轉(zhuǎn)錄因子模塊分析,擬時序分析,RNA 速率分析,基因功能富集分析等,共鑒定了 101 種細(xì)胞類型,包括上皮細(xì)胞和間充質(zhì)祖細(xì)胞群和與關(guān)鍵形態(tài)發(fā)生的重要程序。作者描述了隱窩 - 絨毛軸形成的原理,發(fā)育中的腸道的神經(jīng)、血管、間充質(zhì)形態(tài)發(fā)生和免疫群體。確定了發(fā)育中的成纖維細(xì)胞和肌成纖維細(xì)胞亞型的分化層次,并描述了它們的不同功能,包括作為血管生態(tài)位細(xì)胞的功能。作者確定了 Peyer’s patches 和 gut-associated lymphoid tissue (GALT) 的起源,并描述了位置特異性免疫程序。提出了一個無偏倚的分析形態(tài)梯度,可以用來指引連續(xù)的細(xì)胞分化、定義細(xì)胞、以及罕見的發(fā)育性腸道疾病相關(guān)的位置區(qū)域。此外,作者還編制了一個公開的胎兒腸道在時間和空間層面發(fā)育(STAR-FINDer) 的在線數(shù)據(jù)庫,以促進(jìn)進(jìn)一步的工作。
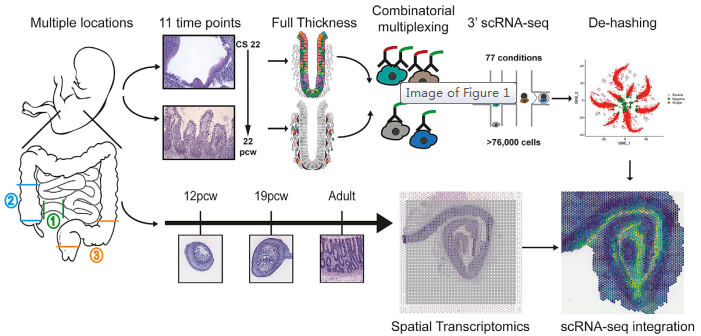
圖 研究概況
案例四
空間轉(zhuǎn)錄組測序揭示阿爾茲海默疾病機(jī)制
Spatial Transcriptomics and In Situ Sequencing to Study Alzheimer’s Disease
發(fā)表期刊:Cell
影響因子:38.637
發(fā)表時間:2020 年 8 月
雖然在阿爾茨海默病(AD) 淀粉樣斑塊周圍觀察到復(fù)雜的炎癥樣改變,但對這種反應(yīng)的分子變化和細(xì)胞相互作用知之甚少。在 AD 小鼠模型中,作者利用空間轉(zhuǎn)錄組學(xué)技術(shù)研究淀粉樣斑塊周圍直徑為 100 毫米的組織結(jié)構(gòu)域發(fā)生的轉(zhuǎn)錄變化。研究證實了髓磷脂和少突膠質(zhì)細(xì)胞基因(OLIGs) 富集的基因共表達(dá)網(wǎng)絡(luò)的早期改變,而涉及補體系統(tǒng)、氧化應(yīng)激、溶酶體和炎癥的斑塊誘導(dǎo)基因(豬)的多細(xì)胞基因共表達(dá)網(wǎng)絡(luò)在疾病的后期顯著。此外,在小鼠和人類大腦切片上使用原位測序確認(rèn)了大多數(shù)在細(xì)胞水平上觀察到的改變。空間轉(zhuǎn)錄組學(xué)分析為解開 AD 和其他腦部疾病致病特征附近的失調(diào)細(xì)胞網(wǎng)絡(luò)提供了一種新的方法。
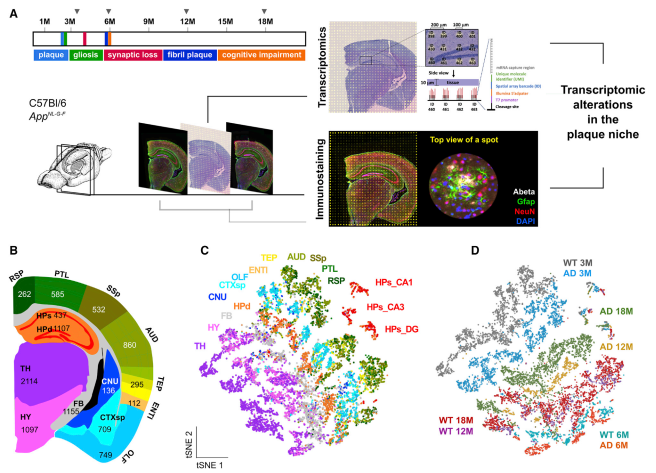
圖 成年 AD 小鼠大腦的空間解析轉(zhuǎn)錄組譜
案例五
空間轉(zhuǎn)錄組圖譜為前列腺癌提供異質(zhì)性的新視圖
Spatial Maps of Prostate Cancer Transcriptomes Reveal an Unexplored
Landscape of Heterogeneity
發(fā)表期刊:Nature communication
影響因子:12.212
發(fā)表時間:2018 年 9 月
前列腺癌包含大量的腫瘤內(nèi)異質(zhì)性,在原發(fā)腫瘤和遠(yuǎn)距離轉(zhuǎn)移中都存在基因改變前列腺癌的病理嚴(yán)重程度,盡管有分子標(biāo)記和核磁共振的進(jìn)展,但通常是根據(jù) Gleason 分級(Gs)系統(tǒng)來評分的,該系統(tǒng)僅使用組織學(xué)數(shù)據(jù),經(jīng)常在血液和腫瘤分期中補充 PSA 測量。然而,這種分類方法有局限性,并提出了新的備選方案。利用一種新的反卷積方法,作者分析了近 6750 個組織區(qū)域的轉(zhuǎn)錄組,并提取了不同組織成分的不同表達(dá)譜,如間質(zhì)、正常和針腺、免疫細(xì)胞和癌癥。同時區(qū)分了健康區(qū)域和病變區(qū)域,從而對前列腺癌進(jìn)展過程中的基因表達(dá)變化提供了見解。與病理學(xué)家的注釋相比,空間轉(zhuǎn)錄組可以更準(zhǔn)確地描述了癌灶的范圍,有趣的是,與組織學(xué)變化無關(guān)。
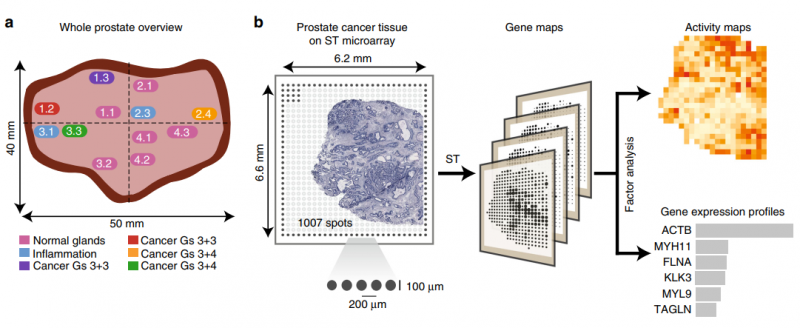
圖 前列腺癌的空間轉(zhuǎn)錄組學(xué)(ST) 研究設(shè)計
案例六
空間轉(zhuǎn)錄組解析模式植物組織基因表達(dá)的空間信息
Spatially resolved transcriptome profiling in model plant species
發(fā)表期刊:Nature plant
影響因子:13.256
發(fā)表時間:2017 年 5 月
要理解復(fù)雜的生物系統(tǒng),需要對特定組織域進(jìn)行功能研究。然而,現(xiàn)有的空間轉(zhuǎn)錄組測序技術(shù)僅適用于有限范圍的生物,主要是哺乳動物。在這里,作者提出了可用與植物組織的方法,在空間分辨率的狀態(tài)下來廣泛的解析模式植物系統(tǒng)。該過程包括高通量的空間轉(zhuǎn)錄組分析,然后是空間基因和通路分析。作者首先從模型被子植物和裸子植物的顯微切片中生成空間轉(zhuǎn)錄組譜,證明了該技術(shù)的可行性。在擬南芥中,利用空間數(shù)據(jù)分析了 8 個花序組織結(jié)構(gòu)域中 141 個基因和 189 條通路的表達(dá)水平差異。作者通過將空間轉(zhuǎn)錄組學(xué)技術(shù)和功能譜分析技術(shù)相結(jié)合這一新策略,應(yīng)用于廣泛的植物物種,該技術(shù)將是一種解決發(fā)育和進(jìn)化生物學(xué)基本問題的關(guān)鍵方法。
▲圖 植物的空間解析轉(zhuǎn)錄組分析

[1]. St?hl PL, Salmén F, Vickovic S, et al. Visualization and analysis of gene expression in tissue sections by spatial transcriptomics. Science 2016, 353(6294):78-82.
[2]. Peng G, Suo S, Chen J, et al. Spatial Transcriptome for the Molecular Annotation of Lineage Fates and Cell Identity in Mid-gastrula Mouse Embryo. Dev Cell 2016, 36(6):681-697.
[3]. https://www.spatialomics.org/SpatialDB/
[4]. Michaela Asp, Stefania Giacomello, et al. A Spatiotemporal Organ-Wide Gene Expression and Cell Atlas of the Developing Human Heart[J]. Cell, 2019, 179, 1647–1660.
[5]. Maniatis S, ?ij? T, Vickovic S, et al. Spatiotemporal dynamics of molecular pathology in
amyotrophic lateral sclerosis[J]. Science, 2019, 364(6435): 89-93.
[6]. Carlberg K, Korotkova M, Larsson L, Catrina AI, Stahl PL, Malmstrom V. Exploring inflammatory signatures in arthritic joint biopsies with Spatial Transcriptomics[J]. Scientific reports. 2019,9(1):18975.
[7]. José, Fernández, Navarro, et al. ST viewer: a tool for analysis and visualization of spatial transcriptomics datasets.[J]. Bioinformatics (Oxford, England), 2019.
[8]. Berglund E , Maaskola J , Schultz N , et al. Spatial maps of prostate cancer transcriptomes reveal an unexplored landscape of heterogeneity[J]. Nature Communications, 2018, 9(1).
[9]. ST Spot Detector: a web-based application for automatic spot and tissue detection for spatial Transcriptomics image datasets[J]. Bioinformatics, 2018.
[10]. Anna L , Natalija G , Tove B , et al. Gene expression profiling of periodontitis-affected gingival tissue by spatial transcriptomics[J]. Scientific Reports, 2018, 8(1):9370.
[11]. Kim T , Hanna E , Jonas M , et al. Spatially resolved transcriptomics enables dissection of genetic heterogeneity in stage III cutaneous malignant melanoma[J]. Cancer Research, 2018:canres.0747.2018.
[12]. Salmen F, Stahl PL, Mollbrink A, Navarro JF, Vickovic S, Frisen J, et al. Barcoded solid-phase RNA capture for Spatial Transcriptomics profiling in mammalian tissue sections[J]. Nature protocols. 2018,13(11):2501-34.
[13]. Giacomello S , Fredrik Salmén, Terebieniec B K , et al. Spatially resolved transcriptome profiling in model plant species[J]. Nature Plants, 2017, 3(6):17061.
[14]. Asp M , Salmén, Fredrik, St?Hl P L , et al. Spatial detection of fetal marker genes expressed at low level in adult human heart tissue[J]. Scientific Reports, 2017, 7(1):12941.
[15]. Stahl P L , Salmen F , Vickovic S , et al. Visualization and analysis of gene expression in tissue sections by spatial transcriptomics[J]. Science, 2016, 353(6294):78-82.
[16]. Vickovic S , St?Hl P L , Salmén, Fredrik, et al. Massive and parallel expression profiling using microarrayed single-cell sequencing[J]. Nature Communications, 2016, 7:13182.
[17]. Anders, Jemt, Fredrik, et al. An automated approach to prepare tissue-derived spatially barcoded RNA-sequencing libraries.[J]. Scientific reports, 2016.
[18] Berglund E , Maaskola J , Schultz N , et al. Spatial maps of prostate cancer transcriptomes reveal an unexplored landscape of heterogeneity[J]. Nature Communications, 2018, 9(1).
[19] Chen W T , Lu A , Craessaerts K , et al. Spatial Transcriptomics and In Situ Sequencing to Study Alzheimer's Disease[J]. Cell, 2020.
[20] Giacomell S , Fredrik Salmén, Terebieniec B K , et al. Spatially resolved transcriptome profiling in model plant species[J]. Nature Plants, 2017.
[21] David Fawkner-Corbett, et al. Spatiotemporal analysis of human intestinal development at single-cell resolution[J]. Cell. 2021 Feb 4;184(3):810-826.e23. doi: 10.1016/j.cell.2020.12.016. Epub 2021 Jan 5.

| 序號 | 文件類型 | 查閱 |
| 1 | 【畫冊】空間基因表達(dá)解決方案 | 點擊查看 |
| 2 | 【畫冊】石蠟樣本(FFPE)空間基因表達(dá)解決方案 | 點擊查看 |
| 3 | 【畫冊】新鮮樣本(FF)空間基因表達(dá)解決方案 | 點擊查看 |

